-
中文名稱:人PKD1重組蛋白
-
品名簡稱:Recombinant Human PKD1 protein, partial
-
貨號:CSB-EP018051HUa0
-
說明書:
-
規格:¥1836
-
圖片:
-
 (Tris-Glycine gel) Discontinuous SDS-PAGE (reduced) with 5% enrichment gel and 15% separation gel.
(Tris-Glycine gel) Discontinuous SDS-PAGE (reduced) with 5% enrichment gel and 15% separation gel.
-
-
其他:
產品詳情
-
純度:Greater than 85% as determined by SDS-PAGE.
-
基因名:PKD1
-
Uniprot No.:
-
別名:PC1;Autosomal dominant polycystic kidney disease 1 protein
-
種屬:Homo sapiens (Human)
-
蛋白長度:Partial
-
來源:E.coli
-
分子量:69.3 kDa
-
表達區域:25-636aa
-
氨基酸序列GPGRGCGPCEPPCLCGPAPGAACRVNCSGRGLRTLGPALRIPADATALDVSHNLLRALDVGLLANLSALAELDISNNKISTLEEGIFANLFNLSEINLSGNPFECDCGLAWLPRWAEEQQVRVVQPEAATCAGPGSLAGQPLLGIPLLDSGCGEEYVACLPDNSSGTVAAVSFSAAHEGLLQPEACSAFCFSTGQGLAALSEQGWCLCGAAQPSSASFACLSLCSGPPPPPAPTCRGPTLLQHVFPASPGATLVGPHGPLASGQLAAFHIAAPLPVTATRWDFGDGSAEVDAAGPAASHRYVLPGRYHVTAVLALGAGSALLGTDVQVEAAPAALELVCPSSVQSDESLDLSIQNRGGSGLEAAYSIVALGEEPARAVHPLCPSDTEIFPGNGHCYRLVVEKAAWLQAQEQCQAWAGAALAMVDSPAVQRFLVSRVTRSLDVWIGFSTVQGVEVGPAPQGEAFSLESCQNWLPGEPHPATAEHCVRLGPTGWCNTDLCSAPHSYVCELQPGGPVQDAENLLVGAPSGDLQGPLTPLAQQDGLSAPHEPVEVMVFPGLRLSREAFLTTAEFGTQELRRPAQLRLQVYRLLSTAGTPENGSEPESRSPDNRTQL
Note: The complete sequence may include tag sequence, target protein sequence, linker sequence and extra sequence that is translated with the protein sequence for the purpose(s) of secretion, stability, solubility, etc.
If the exact amino acid sequence of this recombinant protein is critical to your application, please explicitly request the full and complete sequence of this protein before ordering. -
蛋白標簽:N-terminal 6xHis-tagged
-
產品提供形式:Liquid or Lyophilized powder
Note: We will preferentially ship the format that we have in stock, however, if you have any special requirement for the format, please remark your requirement when placing the order, we will prepare according to your demand. -
緩沖液:If the delivery form is liquid, the default storage buffer is Tris/PBS-based buffer, 5%-50% glycerol. If the delivery form is lyophilized powder, the buffer before lyophilization is Tris/PBS-based buffer, 6% Trehalose.
-
復溶:We recommend that this vial be briefly centrifuged prior to opening to bring the contents to the bottom. Please reconstitute protein in deionized sterile water to a concentration of 0.1-1.0 mg/mL.We recommend to add 5-50% of glycerol (final concentration) and aliquot for long-term storage at -20°C/-80°C. Our default final concentration of glycerol is 50%. Customers could use it as reference.
-
儲存條件:Store at -20°C/-80°C upon receipt, aliquoting is necessary for mutiple use. Avoid repeated freeze-thaw cycles.
-
保質期:The shelf life is related to many factors, storage state, buffer ingredients, storage temperature and the stability of the protein itself.
Generally, the shelf life of liquid form is 6 months at -20°C/-80°C. The shelf life of lyophilized form is 12 months at -20°C/-80°C. -
貨期:3-7 business days
-
注意事項:Repeated freezing and thawing is not recommended. Store working aliquots at 4°C for up to one week.
-
Datasheet & COA:Please contact us to get it.
產品評價

相關產品
靶點詳情
-
功能:Component of a heteromeric calcium-permeable ion channel formed by PKD1 and PKD2 that is activated by interaction between PKD1 and a Wnt family member, such as WNT3A and WNT9B. Both PKD1 and PKD2 are required for channel activity. Involved in renal tubulogenesis. Involved in fluid-flow mechanosensation by the primary cilium in renal epithelium. Acts as a regulator of cilium length, together with PKD2. The dynamic control of cilium length is essential in the regulation of mechanotransductive signaling. The cilium length response creates a negative feedback loop whereby fluid shear-mediated deflection of the primary cilium, which decreases intracellular cAMP, leads to cilium shortening and thus decreases flow-induced signaling. May be an ion-channel regulator. Involved in adhesive protein-protein and protein-carbohydrate interactions.
-
基因功能參考文獻:
- Two novel mutations, c.6953_6977del and c.10937T>G (p.Val3646Gly) of the PKD1 gene are associated with the polycystic kidney disease. PMID: 29896735
- this study reports the 3.6-angstrom cryo-electron microscopy structure of truncated human PKD1-PKD2 complex assembled in a 1:3 ratio. PMID: 30093605
- VEGF/PKD-1 signaling axis increases angiogenic and arteriogenic gene expression. These studies suggest that the axis may regulate arteriolar differentiation through changing microvascular endothelial cells gene expression. PMID: 29380239
- The novel frameshift mutations reported by this study are p. Q1997X, P. D73X and p. V336X. PMID: 29590654
- newly identified sites for known mutations will facilitate the early diagnosis and prediction of prognosis in patients with ADPKD PMID: 29529603
- PKD1 cytoplasmic C-terminal tail domain has a crucial role in renal prognosis in autosomal dominant polycystic disease. PMID: 28983800
- PKD 1 mutation is associated with Autosomal dominant polycystic kidney disease. PMID: 28825164
- Our results show that we have successfully generated a patient-specific iPS cell line with a mutation in PKD1 for study of renal disease pathophysiology. PMID: 29034881
- These data reveal a novel function for PKD1 as a regulator of focal adhesion dynamics and by identifying PIP5Klgamma as a novel PKD1 substrate provide mechanistic insight into this process. PMID: 27775029
- the PKD1/PKD2 mutation status differed by ethnicity, and the PKD1/PKD2 genotype may affect the clinical phenotype of autosomal dominant polycystic kidney disease PMID: 27782177
- The novel pathogenic variants c.3607C> T and c.11354G> C in PKD1 is very interesting since they may represent Italian clusters. PMID: 27499327
- SNX3-retromer complex regulates the surface expression and function of PC1 and PC2 PMID: 28620080
- Study identified a novel heterozygous frameshift mutation in PKD1 gene segregating between affected and unaffected individuals suggesting an involvement in polycystic kidney disease (PKD). PMID: 28870863
- we report for the first time that PKD1 was tightly regulated by androgen at the transcriptional level in prostate cancer cells and was a novel androgen-repressed gene. Further analysis identified FRS2 as a novel mediator of androgen-induced PKD1 repression. PMID: 28077787
- novel frameshift mutation c.3903delC, p.A1302Pfs identified to be responsible for renal disease PMID: 28827396
- Hyperactivation of the ERK pathway may be caused by down-regulation of PC-1 and PC-2 in lymphatic malformations, contributing to increased proliferation of lymphatic endothelial cells. PMID: 28552828
- A novel mutation of the PKD1 gene has been identified with autosomal dominant polycystic kidney disease in an affected Chinese family. PMID: 28604956
- Annualized median liver growth rates were 1.68, 1.5 and 1.24% for PKD1-T, PKD1-NT and PKD2 mutations, respectively (P = 0.49), and remained unaffected by the ADPKD genotype when adjusted for age, gender and baseline HtLV. PMID: 26932689
- this study shows that diet-induced obesity links to estrogen receptor-positive breast cancer progression via LPA/PKD-1-CD36 signaling-mediated microvascular remodeling PMID: 28186980
- Isolated polycystic liver disease genes define effectors of polycystin-1 function PMID: 28375157
- mechanical load upregulates expression of Runx2 gene via potentiation of PC1-JAK2/STAT3 signaling axis, culminating to possibly control osteoblastic differentiation and ultimately bone formation. PMID: 27699453
- Gly972Arg of PC-1 polymorphisms are associated with polycystic ovary syndrome. PMID: 27785750
- cortactin binds to E-cadherin, and that a posttranslational modification of cortactin, RhoA-induced phosphorylation by protein kinase D1 (PKD1; also known as PRKD1) at S298, impairs adherens junction assembly and supports their dissolution. PMID: 27179075
- Ten novel mutations in PKD1 gene were identified in 15 Chinese families with polycystic kidney disease. PMID: 28578020
- The novel PKD1 c.8791+1_8791+5delGTGCG mutation has created a new splice site. PMID: 27984604
- Data show that in 82 (87.2%) of the patients, pathogenic mutations were detected in PKD1 (79.0%-92.5%), whereas in 12 (12.8%) patients pathogenic mutations were detected in PKD2 (7.5%-21.0%). PMID: 27835667
- our study reveals possible mechanisms of CFB upregulation in autosomal dominant polycystic kidney disease, and a novel role of PC1-C terminal tail in ADPKD-associated inflammation. PMID: 26984954
- PKD1 mutation is associated with familial testicular germ cell tumor and autosomal dominant polycystic kidney disease. PMID: 27577987
- we have quantified the contribution of genic and PKD1 allelic effects and sex to the Autosomal dominant polycystic kidney disease phenotype. Intrafamilial correlation analysis showed that other factors shared by families influence height-adjusted total kidney volume, with these additional genetic/environmental factors significantly affecting the Autosomal dominant polycystic kidney disease phenotype PMID: 26823553
- The epithelial cell membrane protein E-cadherin physically binds to PKD1 which leads to a subcellular redistribution of PKD1. PMID: 26991955
- PKD1 and PKD2 are susceptibility genes for familial intracranial aneurysms in a Japanese population. PMID: 27567292
- These results suggest that the Ser at the 4166 site in PC1 is crucial in the PC1 mediated MEK/ERK/myc signaling pathway, which might be the key pathophysiological cause of Aortic dissection. PMID: 28076932
- Novel PKD1 mutations in Chinese autosomal dominant polycystic kidney disease patients PMID: 26632257
- The pathogenic mutation in PKD1 linked pedigree was c.8522G>A (p.E2771K) in exon 23. This C to T transition occurs at the CpG dinucleotides which is the known hotspot point for mutations. PMID: 26950445
- Germline mutations in PKD1 gene is associated with autosomal-dominant polycystic kidney disease. PMID: 23300259
- Host cortactin, PKD1 and actin are recruited by Trypanosoma cruzi extracellular amastigotes based on experiments in fixed and live cells by time lapse confocal microscopy. PMID: 26096820
- PCs expression and p53 activation as a regulator of cell proliferation were further evaluated in vivo and in 69 advanced human carotid atherosclerotic plaques. PMID: 26286632
- These results suggest that, at least in some patients, the severity of the cystic disease is inversely correlated with the level of polycystin 1 function. PMID: 26139440
- Overexpression of PKD1 in a prostate cancer cell line model resulted in decreased cell proliferation and epithelial mesenchymal transition. PMID: 26764245
- and MMP9 expression in PKD1 constitutively-active MD-MB-231 cells and MCF-7 knockdown cells were decreased and increased respectively PMID: 26775353
- A substantial number of PKD1 missense or synonymous mutations characterize pre-mRNA splicing. One missense and 2 synonymous mutations induce significant defects in pre-mRNA splicing. PMID: 25757501
- PKD1 gene variation plays a disease modifying role in patients diagnosed with ADPKD. PMID: 25880449
- A short segment of chromosome 16 encodes the tumor suppressor gene tuberin as well as the protein polycystin 1 which are responsible for tuberous sclerosis complex type 2 and autosomal-dominant polycystic kidney disease type 1, respectively. PMID: 25355409
- Case Report: polycystic kidney disease with steatocystoma multiplex. PKD1 mutations disrupt keratin 17 polymerization. PMID: 25111597
- mutations within PKD1 and PKD2 can be linked to most of the cases of Japanese ADPKD, and the renal function decline was faster in patients with PKD1 mutations than in those with PKD2 mutations PMID: 24611717
- PKD1 knockdown was found to be an efficient strategy to overcome resistance caused by LMP1 expression. Therefore, PKD1 could be a molecular target for therapeutic intervention in EBV-associated B cell lymphoma treatment. PMID: 24707946
- Report novel nonsense mutation in exon 15 of PKD1 in polycystic liver disease. PMID: 25741140
- Four novel PKD1 variants were identified in this study in autosomal-dominant polycystic kidney disease using long-range PCR followed by nested PCR PMID: 25531466
- Data indicate that serine/threonine-protein kinase D1 (PKD1) stimulates estrogen receptor alpha (ERalpha) expression in breast cancer MCF-7 cells. PMID: 25287328
- Both polycystins were detected on the spindle and mid-body of mitotic cells, while fibrocystin was on centrosome throughout cell cycle. PMID: 25367197
顯示更多
收起更多
-
相關疾病:Polycystic kidney disease 1 (PKD1)
-
亞細胞定位:Cell membrane; Multi-pass membrane protein. Cell projection, cilium. Endoplasmic reticulum. Golgi apparatus.
-
蛋白家族:Polycystin family
-
數據庫鏈接:
Most popular with customers
-
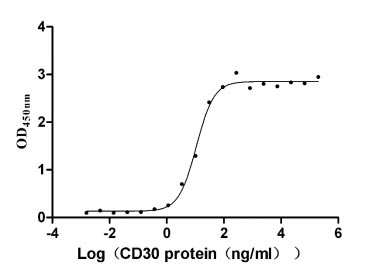
Recombinant Human Tumor necrosis factor ligand superfamily member 8 (TNFSF8), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
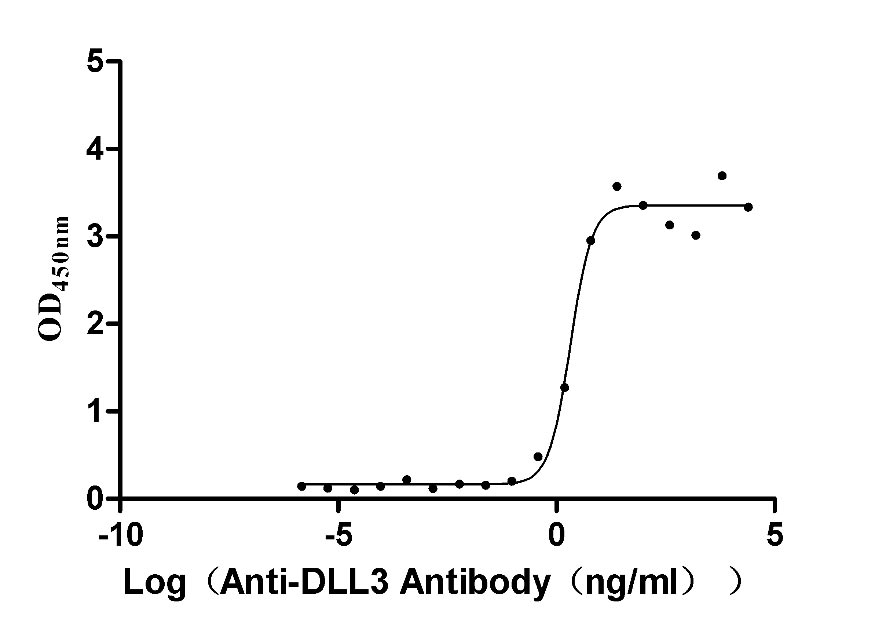
Recombinant Macaca fascicularis Delta-like protein 3 (DLL3), partial (Active)
Express system: Mammalian cell
Species: Macaca fascicularis (Crab-eating macaque) (Cynomolgus monkey)
-
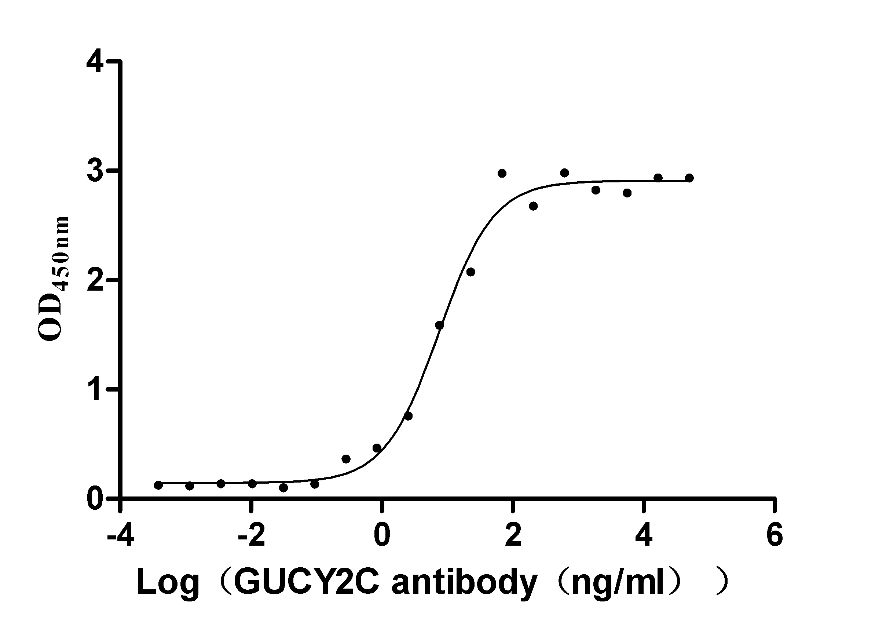
Recombinant Human Heat-stable enterotoxin receptor (GUCY2C), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
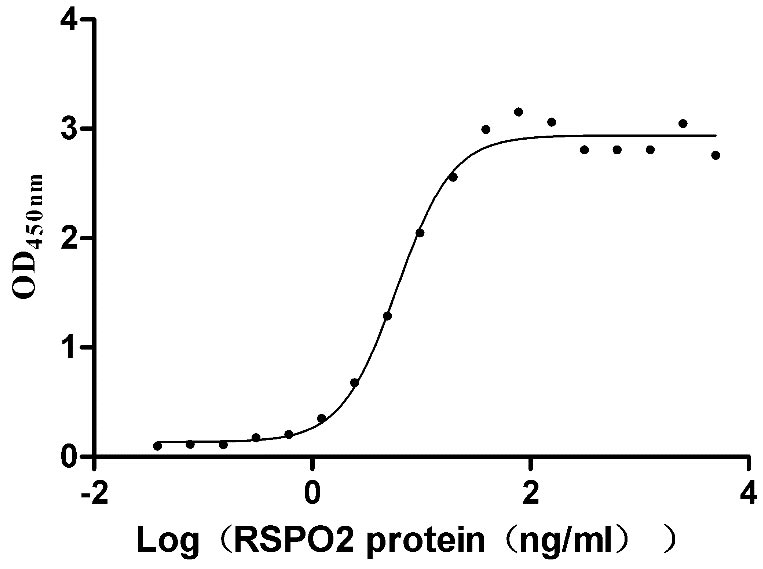
Recombinant Human E3 ubiquitin-protein ligase ZNRF3 (ZNRF3), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
Recombinant Macaca fascicularis CD93 molecule (CD93), partial (Active)
Express system: Mammalian cell
Species: Macaca fascicularis (Crab-eating macaque) (Cynomolgus monkey)
-
Recombinant Macaca fascicularis Trophoblast glycoprotein (TPBG), partial (Active)
Express system: Mammalian cell
Species: Macaca fascicularis (Crab-eating macaque) (Cynomolgus monkey)
-
Recombinant Macaca fascicularis zymogen granule protein 16 homolog B (ZG16B) (Active)
Express system: Mammalian cell
Species: Macaca fascicularis (Crab-eating macaque) (Cynomolgus monkey)
-
Recombinant Human Alkaline phosphatase, germ cell type (ALPG) (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)















