Recombinant Human Peripheral myelin protein 22 (PMP22), partial
-
中文名稱:人PMP22重組蛋白
-
貨號:CSB-YP018241HU1
-
規格:
-
來源:Yeast
-
其他:
-
中文名稱:人PMP22重組蛋白
-
貨號:CSB-EP018241HU1
-
規格:
-
來源:E.coli
-
其他:
-
中文名稱:人PMP22重組蛋白
-
貨號:CSB-EP018241HU1-B
-
規格:
-
來源:E.coli
-
共軛:Avi-tag Biotinylated
E. coli biotin ligase (BirA) is highly specific in covalently attaching biotin to the 15 amino acid AviTag peptide. This recombinant protein was biotinylated in vivo by AviTag-BirA technology, which method is BriA catalyzes amide linkage between the biotin and the specific lysine of the AviTag.
-
其他:
-
中文名稱:人PMP22重組蛋白
-
貨號:CSB-BP018241HU1
-
規格:
-
來源:Baculovirus
-
其他:
-
中文名稱:人PMP22重組蛋白
-
貨號:CSB-MP018241HU1
-
規格:
-
來源:Mammalian cell
-
其他:
產品詳情
-
純度:>85% (SDS-PAGE)
-
基因名:
-
Uniprot No.:
-
別名:PMP22; GAS3; Peripheral myelin protein 22; PMP-22; Growth arrest-specific protein 3; GAS-3
-
種屬:Homo sapiens (Human)
-
蛋白長度:Partial
-
蛋白標簽:Tag?type?will?be?determined?during?the?manufacturing?process.
The tag type will be determined during production process. If you have specified tag type, please tell us and we will develop the specified tag preferentially. -
產品提供形式:Lyophilized powder
Note: We will preferentially ship the format that we have in stock, however, if you have any special requirement for the format, please remark your requirement when placing the order, we will prepare according to your demand. -
復溶:We recommend that this vial be briefly centrifuged prior to opening to bring the contents to the bottom. Please reconstitute protein in deionized sterile water to a concentration of 0.1-1.0 mg/mL.We recommend to add 5-50% of glycerol (final concentration) and aliquot for long-term storage at -20℃/-80℃. Our default final concentration of glycerol is 50%. Customers could use it as reference.
-
儲存條件:Store at -20°C/-80°C upon receipt, aliquoting is necessary for mutiple use. Avoid repeated freeze-thaw cycles.
-
保質期:The shelf life is related to many factors, storage state, buffer ingredients, storage temperature and the stability of the protein itself.
Generally, the shelf life of liquid form is 6 months at -20°C/-80°C. The shelf life of lyophilized form is 12 months at -20°C/-80°C. -
貨期:Delivery time may differ from different purchasing way or location, please kindly consult your local distributors for specific delivery time.Note: All of our proteins are default shipped with normal blue ice packs, if you request to ship with dry ice, please communicate with us in advance and extra fees will be charged.
-
注意事項:Repeated freezing and thawing is not recommended. Store working aliquots at 4°C for up to one week.
-
Datasheet :Please contact us to get it.
產品評價

相關產品
靶點詳情
-
功能:Might be involved in growth regulation, and in myelinization in the peripheral nervous system.
-
基因功能參考文獻:
- This study supported the notion that missense mutations in PMP22 give rise to a Charcot-Marie-Tooth Disease phenotype, possibly through a toxic gain-of-function mechanism. PMID: 28748849
- We identified that PMP22 not only acts as a marker for gastric CSCs but may also have an essential role in regulating the self-renewal and chemoresistance of gastric cancer. Our findings suggest that PMP22 has clinical value for the prognosis and treatment of chemoresistant gastric cancer PMID: 28336807
- In this Chinese Han population, the frequency of PMP22 gene duplication in those with CMT1 was slightly (50% vs. 70%-80%) less than in Western/Caucasian populations. PMID: 27862672
- PMP22 polymorphism is associated with tuberculosis. PMID: 27623071
- The studies implicating GAS3 protein family (EMP1, EMP2, EMP3 and PMP22) in cancer pathogenesis as well as probe the structural similarities between the family members were highlighted. PMID: 27279240
- A Computational Approach to Identify a Potential Alternative Drug With Its Positive Impact Toward PMP22. PMID: 28374912
- Exome sequencing identified MFN2 SNVs in two of the individuals. Neuropathy-associated CNV outside of the PMP22 locus is rare in Charcot-Marie-Tooth (CMT) disease . Nevertheless, there is potential clinical utility in testing for CNVs and exome sequencing in CMT cases negative for the CMT1A duplication. PMID: 26378787
- We discovered that Tead1 and co-activators Yap and Taz are required for Pmp22 expression, as well as for the expression of Egr2 Tead1 directly binds Pmp22 and Egr2 enhancers early in development and Tead1 binding is induced during myelination, correlating with Pmp22 expression. The data identify Tead1 as a novel regulator of Pmp22 expression during development in concert with Sox10 and Egr2 PMID: 27288457
- This study demonstrated We show that blink reflex studies are reliable for identification of inherited demyelinating polyneuropathy (with pmp22 mutation) regardless of severity and can facilitate algorithmic decisions in genetic testing. PMID: 27422849
- Findings suggest that miR-200bc/429 inhibit OS cells proliferation and invasion by targeting PMP22, and function as a tumor suppressor. PMID: 28234890
- PMP22 deletion leads to functional, metabolic and macro-structural alterations in the afferent visual system of hereditary neuropathy with liability to pressure palsies patients. PMID: 27749933
- we report molecular and clinical characterizations of six subjects with the reciprocal phenomenon of deletions spanning both genes, i.e., PMP22-RAI1 deletions. Systematic clinical studies revealed features consistent with SMS, including features of intellectual disability, speech and gross motor delays, behavioral problems and ocular abnormalities. PMID: 27386852
- Data suggest that the father has carried the same duplication of the peripheral myelin protein 22 (PMP22) gene but with no detectable symptom may be due to irregular transmission pattern of the mutation. PMID: 27577214
- our data suggest that an alteration of mRNA processing could be a pathogenic mechanism in CMT1A. PMID: 26486801
- These results suggest that the severe congenital hypomyelinating neuropathy that characterizes Tr(J)mice results in structural and functional deficits of the developing Neuromuscular Junction. PMID: 26921370
- Data (including data from studies using recombinant proteins that lack typical in-vivo post-translational modifications such as palmitoylation) suggest PMP22 exhibits little tendency to partition into liquid-ordered domains of unilamellar vesicles. PMID: 26859249
- PMP22 gene knockdown inhibited progression of Chronic Myeloid Leukemia. PMID: 26629937
- The common 17p deletion accounts for approximately 50% and PMP22 micromutations for approximately 2% of cases in a large consecutive cohort of Greek patients with suspected HNPP. PMID: 26110377
- This finding provides compelling evidence that the effects of these mutations on the energetics of PMP22 folding lie at the heart of the molecular basis of Charcot-Marie-Tooth disease. PMID: 26102530
- DNA diagnosis was performed in 5 families with hereditary neuropathy with liability to pressure palsies - the PMP22 deletion was found in 9 patients. PMID: 26977628
- Osteosarcoma metastasis-related gene PMP22 participates in the proliferation, invasion, migration and colony formation of osteosarcoma cells possibly via the MAPK signal transduction pathway PMID: 26154129
- PMP22 is a transmembrane glycoprotein component of myelin, important for myelin functioning. Mutation of PMP22 gene cause Charcot-Marie-Tooth Disease. PMID: 26076881
- PMP22 Gene Duplication is associated with Charcot-Marie-Tooth disease type 1A. PMID: 26544804
- The results of this study revealed distinct patterns of SNP allele frequency distribution, which reliably differentiated CMT1A patients from controls PMID: 24830919
- Charcot-Marie-Tooth disease-related PMP22 is trapped in the endoplasmic reticulum by calnexin-dependent retention and Rer1-mediated early Golgi retrieval systems and partly degraded by the Hrd1-mediated endoplasmic reticulum-associated degradation system. PMID: 25385046
- 2 of 38 CMT2 patients showed rearrangements in the PMP22 gene, which is commonly associated with CMT1 or HNPP phenotypes thus usually not tested in CMT2 patients. PMID: 24819634
- The SNP array has wide coverage, high sensitivity, and high resolution and can be used as a screening tool to detect PMP22 dup/del as shown in this Chinese Han population. PMID: 25522693
- Data suggest that several peripheral myelin protein 22 (PMP22) mutations known to result in neuropathy act by disrupting transmembrane helix packing interactions. PMID: 25243937
- Duplication of PMP22 was detected in 79 of 157 Mexican Charcot-Marie-Tooth Type 1A disease patients. In Mexican patients CMT1A frequency was similar to other populations such as United States, Australia, Finland, Sweden and Spain. PMID: 25030070
- Diagnosis is confirmed by finding respectively a PMP22 duplication, deletion or point mutation PMID: 24646194
- ascorbic acid does not impact on PMP22 transcriptional regulation and PMP22 is not a suitable biomarker for CMT1A PMID: 24812204
- G3BP1 regulation of cell proliferation in breast cancer cells, may occur via a regulatory effect on PMP22 expression. PMID: 24321297
- a novel MDA-7/IL-24-GAS3-beta1integrin-fibronectin signaling pathway that suppresses breast cancer growth, is reported. PMID: 23468528
- This study demonistrated that PMP22 duplication was the most common genetic cause Charcot-Marie-Tooth disease. PMID: 23743332
- Two Brazilian siblings with proptosis were found to have a point mutation in the PMP22 gene. PMID: 23689413
- This review shows that alterations of PM22 levels by mutations of the PM22 gene are responsible for > 50% of all inherited peripheral neuropathies. PMID: 23224996
- Wild type PMP22 possesses minimal conformational stability in micelles, which parallels its poor folding efficiency in the endoplasmic reticulum. PMID: 23639031
- molecular genetic analysis of patients with Bell's palsy failed to demonstrate the presence of the PMP22 gene deletion on chromosome 17q11.2-12 PMID: 23635862
- This study demonistrated that pmp22 gene duplication is the most frequent in patient with Charcot-Marie-Tooth disease in spain. PMID: 24078732
- We describe a novel heterozygous p.Trp39Cys missense mutation in the extracellular domain of the peripheral myelin protein 22 (PMP22) associated with an early-onset demyelinating Charcot-Marie-Tooth type 1 E PMID: 23279344
- altered PMP22 gene expression induces significant central nervous system alterations in patients with hereditary neuropathy with liability to pressure palsies and Charcot-Marie-Tooth 1A PMID: 23243264
- A subset of EGR2 and SOX10 consensus sequences are essential for enhancer activity of the PMP22 gene. PMID: 22180461
- [review] Pes cavus is an early and age-dependent manifestation of CMT1A duplication. PMID: 21590514
- our results suggest a low relative CMT1A duplication frequency in the Greek population PMID: 22243284
- The lower prevalence of HNPP in our study of Finnish conscripts compared to the general population is probably due to pre-service screening. PMID: 21429232
- PMP22 mutation has a role in Charcot-Marie-Tooth disease; the spectrum and frequency of PMP22 mutations in the Slovak population is comparable to that seen in the global population PMID: 22131320
- gene causing Charcot-Marie-Tooth disease (CMT) in the family is found in the 17p11.2-p12 region containing PMP22 gene duplication mutation, resulting in the subtype CMT1A PMID: 22000434
- Four novel point mutations in PMP22 with two different phenotypes: mutation p.Ser79Thr is found in the Dejerine-Sottas neuropathy phenotype. PMID: 22006697
- Structural basis for the Trembler-J phenotype of Charcot-Marie-Tooth disease, the PMP22 Leu16Pro mutation profoundly disrupts the first transmembrane segment destabilizing the protein PMID: 21827951
- More than three-fourths of the patients with Gly94fsX222 mutation demonstrated a CMT1 phenotype combined with transient deficits PMID: 21692910
顯示更多
收起更多
-
相關疾病:Charcot-Marie-Tooth disease 1A (CMT1A); Dejerine-Sottas syndrome (DSS); Hereditary neuropathy with liability to pressure palsies (HNPP); Charcot-Marie-Tooth disease 1E (CMT1E); Inflammatory demyelinating polyneuropathy (IDP)
-
亞細胞定位:Cell membrane; Multi-pass membrane protein.
-
蛋白家族:PMP-22/EMP/MP20 family
-
數據庫鏈接:
Most popular with customers
-
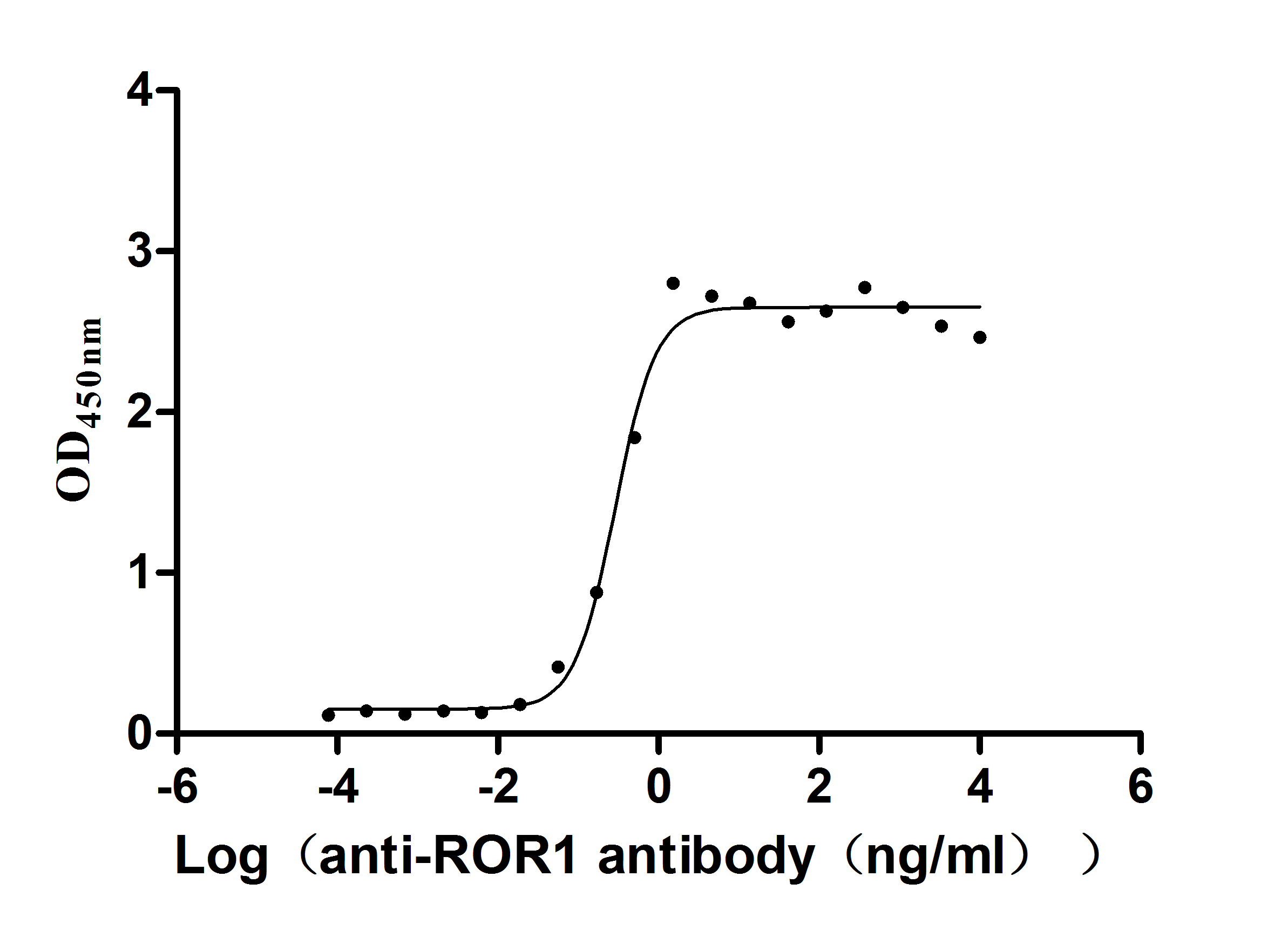
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
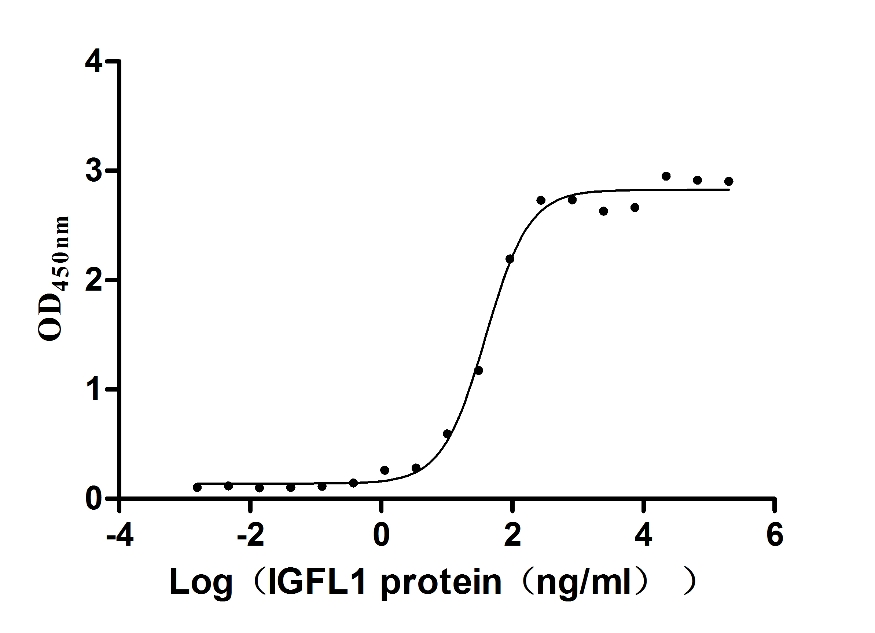
Recombinant Human Insulin growth factor-like family member 1 (IGFL1) (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
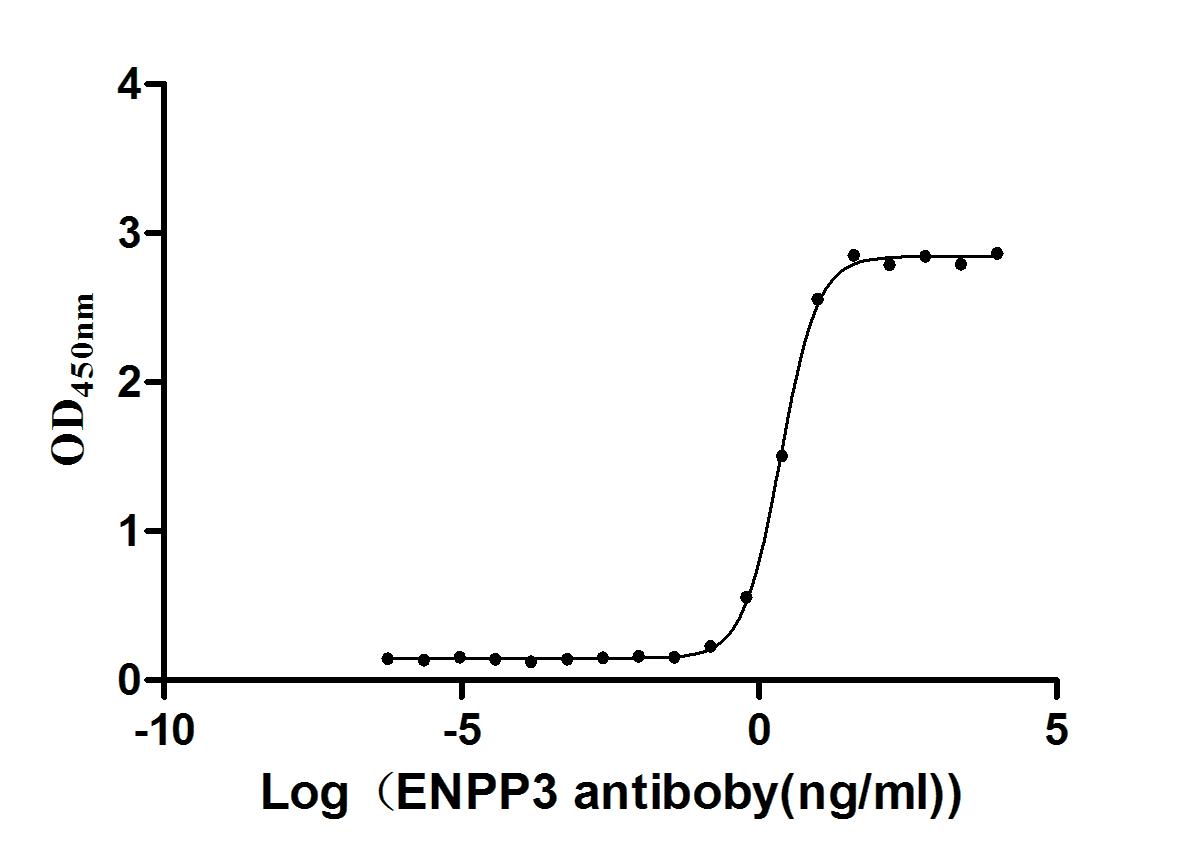
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
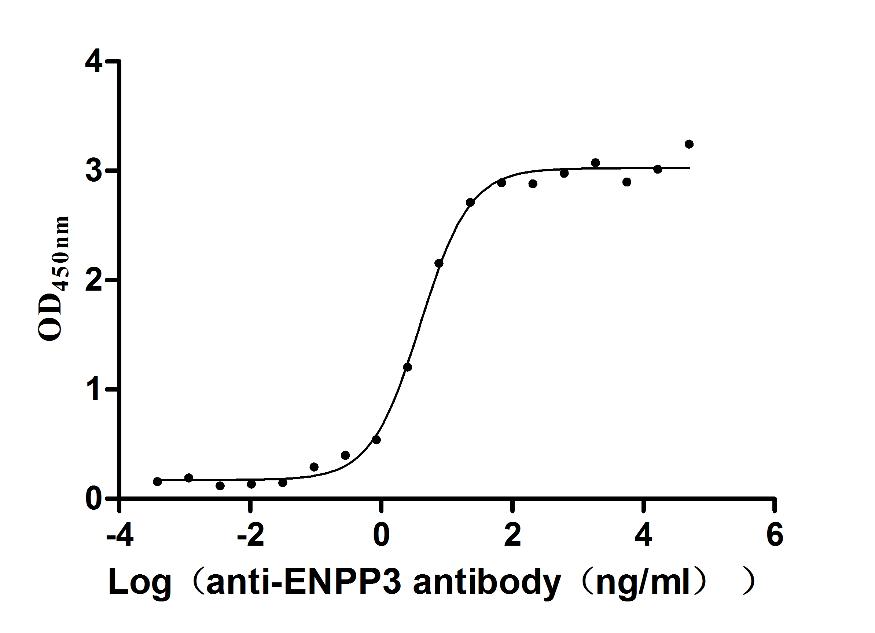
Express system: Mammalian cell
Species: Macaca fascicularis (Crab-eating macaque) (Cynomolgus monkey)
-
Recombinant Macaca fascicularis Membrane spanning 4-domains A1 (MS4A1)-VLPs (Active)
Express system: Mammalian cell
Species: Macaca fascicularis (Crab-eating macaque) (Cynomolgus monkey)
-
Recombinant Human Serine/threonine-protein kinase receptor R3 (ACVRL1), partial (Active)
Express system: Baculovirus
Species: Homo sapiens (Human)
-
Recombinant Human CD70 antigen (CD70), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
Recombinant Human C-C chemokine receptor type 5 (CCR5)-VLPs (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)















