【第87期】前沿靶點速遞:每周醫學研究精選
日期:2026-05-26 11:05:09
01、靶點:FBXO44
應用:胃癌等疾病的潛在治療靶點
來源:MTSS1-dependent ubiquitin modifications mediated by FBXO44 remodel the actin cytoskeleton to promote gastric cancer progression.Mol Cancer,2026 Apr 17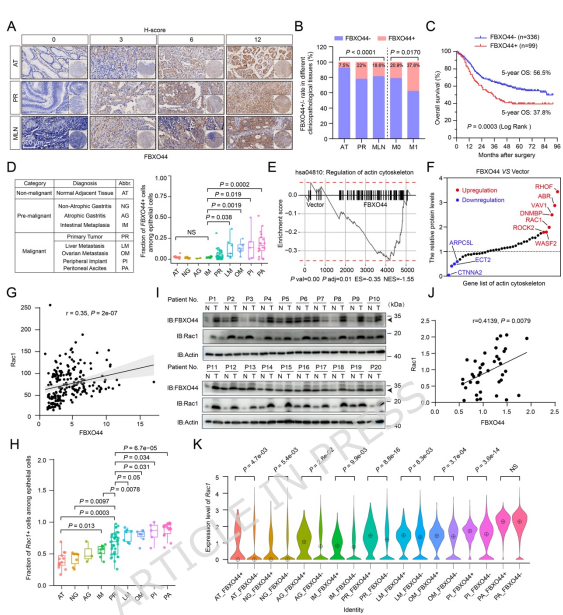
中國科學院杭州醫學研究所與浙江省腫瘤醫院覃江江研究員團隊在Molecular Cancer期刊發表研究論文,揭示FBXO44/MTSS1信號軸介導肌動蛋白細胞骨架重塑促進胃癌進展的新機制。研究發現FBXO44通過與MTSS1相互作用調控Rac1核-胞質轉運,通過K63和K11兩種不同的泛素化程序分別促進Rac1入核和誘導MTSS1降解,這種協調機制重塑Rac1信號分布并介導肌動蛋白細胞骨架重構。FBXO44過表達與晚期胃癌Rac1信號增強相關,FBXO44/MTSS1軸表達平衡顯著影響患者預后。該研究為開發新型抗胃癌藥物提供理論基礎,也為靶向微絲細胞骨架的抗腫瘤治療策略提供新靶點。
02、靶點:PI16
應用:HIV治療的潛在靶點
來源:Systematic discovery of pro- and anti-HIV host factors in primary human CD4+ T cells.Cell,2026 Apr 20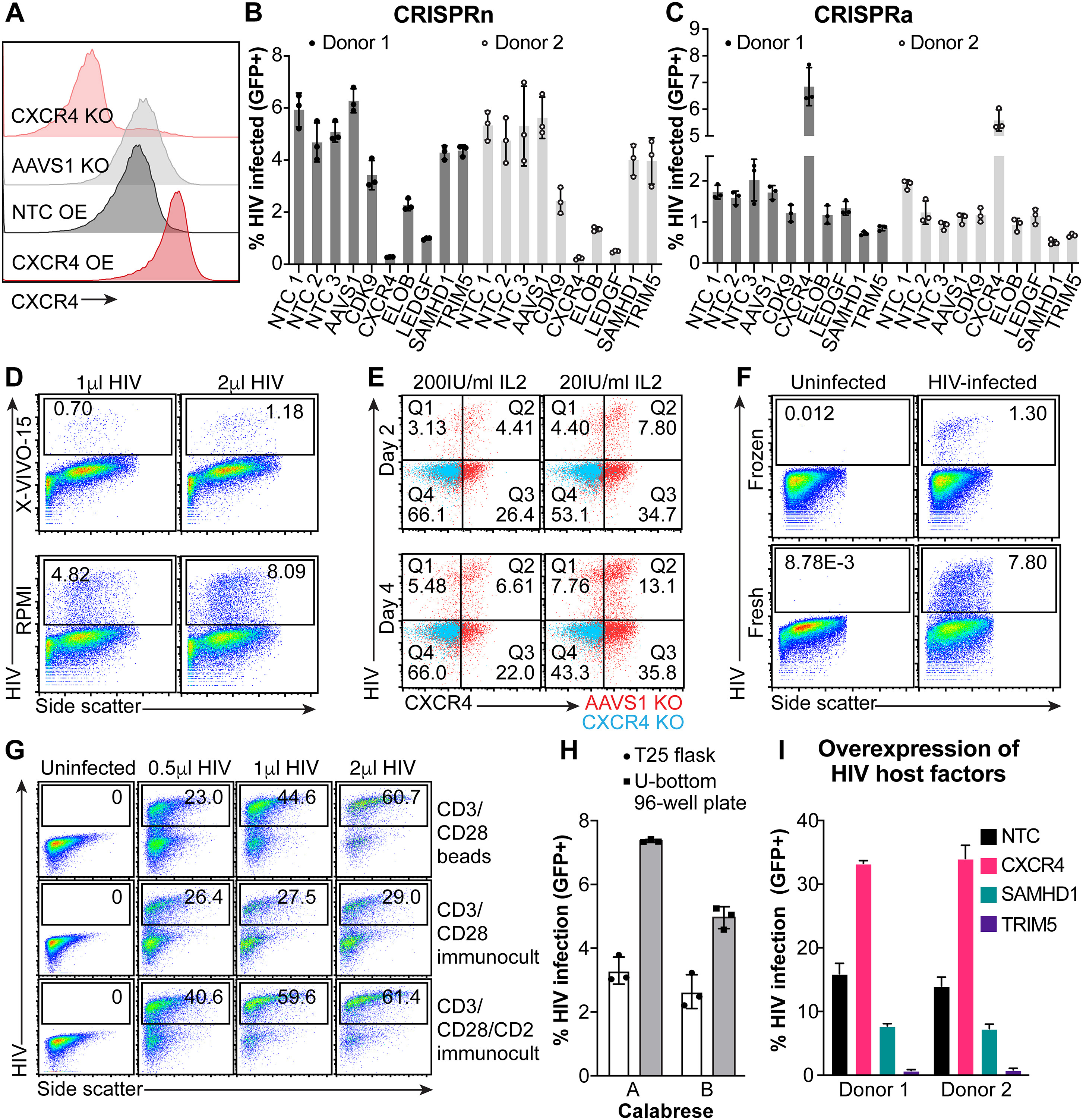
Cell近期發表了一篇研究,研究人員在原代人類CD4+ T細胞中通過CRISPRn和CRISPRa全基因組篩選,系統性發現了決定HIV感染命運的宿主因子。研究揭示PI16通過結合Arp2/3復合物和Gaq信號蛋白抑制病毒包膜與細胞膜融合;親環素家族蛋白PPID(Cyp40)與已知的HIV"幫兇"CypA具有62%序列同一性,但PPID通過特有的TPR結構域招募TOM70等宿主分子機器,嚴重阻礙HIV核心的核輸入。研究發現包括H60P、E248K和H146Y在內的多個氨基酸替換可顯著提升人類PPID的抗病毒活性,構建的CypA-CTPR嵌合體也能將促病毒因子CypA改造為強效抗病毒武器。這些發現為開發基于宿主的HIV干預策略提供了重要靶點。
03、靶點:BRD4
應用:
治療肥胖和胰島素抵抗的潛在靶點
來源:BRD4 directs myofiber identity and metabolic adaptation through CHD4 cooperation.Nat Commun,2026 Apr 09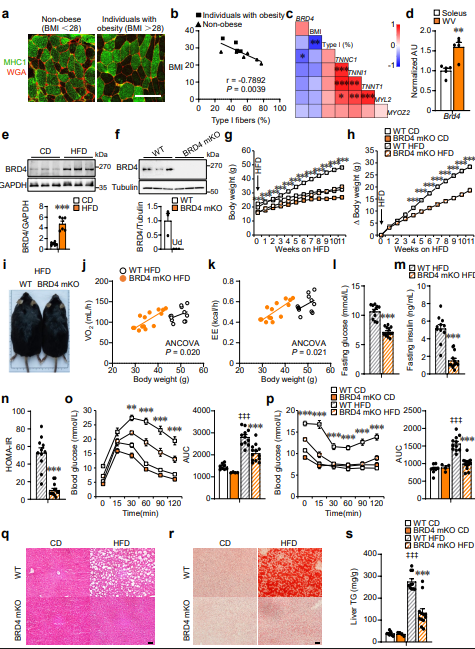
南京大學甘振繼教授團隊在Nature Communications發表研究,揭示表觀遺傳"閱讀器"BRD4作為連接營養信號與骨骼肌代謝表型的關鍵樞紐。研究發現BRD4表達水平與受試者BMI呈正相關、與慢肌纖維比例呈負相關,高脂飲食會導致骨骼肌中BRD4蛋白水平顯著上升。骨骼肌特異性敲除BRD4會誘發"快肌向慢肌"重塑,大幅提升線粒體氧化能力和產熱激活,增加全身能量消耗以抵抗肥胖和胰島素抵抗。機制上,BRD4定位結合快肌基因增強子區域,與MEF2及CHD4形成轉錄調控復合物共同維持快肌基因表達。使用BRD4小分子抑制劑JQ1治療不僅顯著降低脂肪含量、改善糖穩態,更重要的是有效保留肌肉質量,為克服GLP-1類藥物伴隨肌肉萎縮的副作用提供了"減脂保肌"的新策略。
04、靶點:SHANK3
應用:自閉癥及其他神經發育障礙的治療靶點
來源:Dynamic recruitment of CaMKII into SHANK3 phase-separated condensates tunes postsynaptic density remodeling during long-term potentiation.Nat Commun,2026 Apr 20
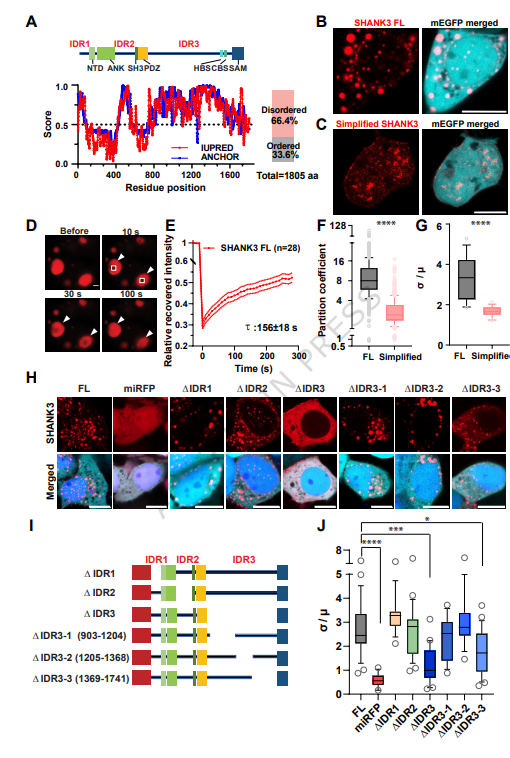
中國科學院深圳先進技術研究院張淑雯團隊聯合黃良宇團隊在Nature Communications發表研究,通過液-液相分離概念揭示自閉癥相關SHANK3蛋白調控突觸可塑性的新機制。研究發現SHANK3通過內在無序區域IDR3(尤其是IDR3-3亞區)介導液相分離,全長SHANK3能形成動態凝聚體并強定位至樹突棘,維持靜息態中正常PSD體積和AMPAR表達。突觸激活后,LTP關鍵激酶CaMKII被迅速招募到SHANK3相分離凝聚體中,該過程需要CaMKII自身磷酸化及SHANK3的IDR3-1亞區內RRK基序,進一步加速凝聚體融合與PSD重塑。與人類自閉癥相關的SHANK3突變(InsG3680)導致部分IDR3及SAM結構域缺失,抑制液相分離功能,造成PSD重塑和AMPAR招募缺陷。該研究揭示了活動依賴性神經可塑性的全新分子機制,為自閉癥及其他神經發育障礙的治療靶點提供了新方向。
05、靶點:TLR8
應用:類風濕性關節炎的潛在治療靶點
來源:Toll-like receptor 8 is a therapeutic target in rheumatoid arthritis.Arthritis Rheumatol,2026 Apr 13
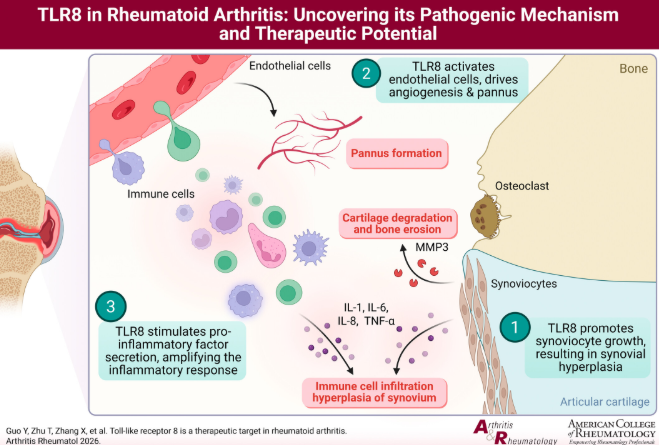
清華大學尹航課題組聯合協和醫院、北京拓領博泰生物科技有限公司在風濕病學國際期刊Arthritis & Rheumatology發表研究,發現Toll樣受體8(TLR8)是類風濕性關節炎(RA)的關鍵藥物靶點。類風濕性關節炎是高發的慢性自身免疫疾病,2025年全球患病人數約4220萬人,其中中國患者約620萬,目前尚無根治手段。研究團隊發現RA患者體內TLR8高表達且與病情呈正相關性,TLR8通過3條主要途徑推動RA進展:刺激滑膜細胞異常增殖、促進內皮細胞形成新生血管、促使滑膜細胞和免疫細胞釋放更多炎性因子。在臨床前動物模型中,使用TLR8特異性抑制劑進行干預,關節炎臨床評分和腫脹程度顯著下降,關節病理損傷明顯改善。基于該機理開發的TLR8特異性小分子抑制劑目前已進入II期臨床階段并取得優異療效數據,為RA精準治療提供了全新策略。
06、靶點:AFABP/FABP4
應用:治療肥胖相關炎癥性腸病(IBD)的潛在靶點
來源:Obesity impairs gut repair via AFABP-mediated iron overload in intestinal stem cells.Nat Metab,2026 Jan
四川大學華西醫院陳海洋教授團隊在Nature Metabolism發表研究,揭示了肥胖損害腸道修復的全新分子機制。研究發現,肥胖時脂肪細胞分泌的脂肪細胞脂肪酸結合蛋白(AFABP/FABP4)會結合血漿轉鐵蛋白(Transferrin, TSF),干擾鐵轉運功能,導致血清非轉鐵蛋白結合鐵(NTBI)升高,引發腸干細胞鐵過載。鐵過載抑制PPARα信號,導致損傷后過氧化物酶體功能下降,進一步擾亂Wnt/Notch分化相關信號平衡,最終阻斷成熟上皮分化,拖慢腸道修復。研究團隊通過Cre依賴AAV在脂肪組織中定點提高AFABP,直接在瘦鼠身上復現了相關表型,為"脂肪-腸道"器官間通訊提供了強有力的因果證據。相反,脂肪特異性敲除AFABP或使用小分子抑制劑BMS309403處理肥胖小鼠,腸道再生能力顯著恢復。基于AFABP-Transferrin-鐵-過氧化物酶體軸,使用鐵螯合劑DFO或過氧化物酶體/PPAR激活劑(如非諾貝特、NaPB或吡格列酮),均可有效逆轉肥胖小鼠的腸道修復缺陷,為治療肥胖相關炎癥性腸病(IBD)提供了極具潛力的可成藥策略。
07、靶點:BDNF
應用:肌萎縮側索硬化癥(ALS,漸凍癥)的潛在治療靶點
來源:BDNF insufficiency exacerbates ALS progression.Cell Rep Med,
2026 May 19
清華大學魯白教授、復旦大學腦科學轉化研究院袁鵬青年研究員、北京大學第三醫院樊東升教授作為共同通訊作者在Cell子刊Cell Reports Medicine發表研究,證實腦源性神經營養因子(BDNF)不足會加劇肌萎縮側索硬化癥(ALS,漸凍癥)的進展。研究發現,人類BDNF基因中常見的單核苷酸多態性rs6265導致Val66Met突變,降低BDNF分泌量,從而縮短ALS患者的生存期。利用ALS致病基因FUS R521C敲入小鼠模型進一步證明,BDNF半劑量不足會導致壽命縮短、運動功能障礙加速以及運動神經元死亡加劇。重要的是,使用激動性抗體激活BDNF受體TrkB,能夠有效挽救ALS相關表型,在其他ALS小鼠模型中,TrkB激活抗體也顯示出優于目前ALS治療藥物利魯唑的治療效果。該研究表明BDNF不足可能是ALS進展的關鍵促成因素,激活BDNF-TrkB通路代表了一種有前景的ALS治療新策略。
08、靶點:KCNJ2
應用:慢性氣道疾病的潛在治療靶點
來源:KCNJ2 is required for NLRP3 inflammasome activation that drives allergic airway inflammation and remodeling.Adv Sci (Weinh),2026 May
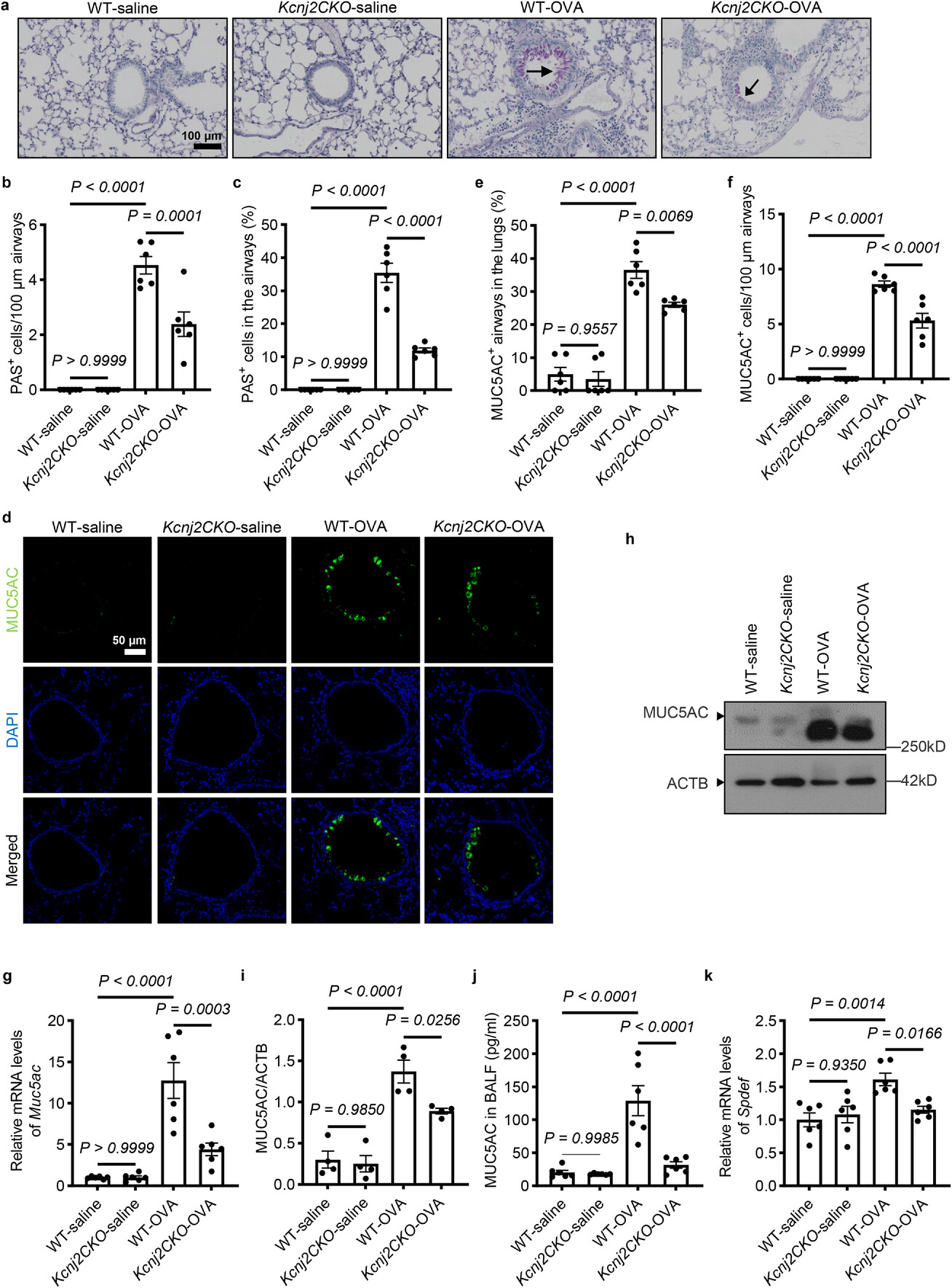
廣州國家實驗室與廣州醫科大學殷文廣、冉丕鑫、李時悅團隊在Advanced Science期刊發表研究,鑒定出慢性氣道疾病新干預靶點KCNJ2,闡明了其驅動2型炎癥反應與氣道重塑的分子機制。2型炎癥為慢性氣道疾病一主要內型,約20%-40%慢阻肺患者、50%-70%哮喘患者與2型炎癥有關。內向整流型鉀離子通道作為鉀離子通道家族中的主要類型,在穩定細胞膜電位中發揮主要作用。研究從臨床患者數據與樣本分析出發,結合動物模型、細胞、類器官培養等,發現了KCNJ2在慢性氣道疾病發生中的作用,提出了"鉀離子通道-炎癥小體-警報素-2型炎癥反應"信號軸調控模型,揭示了其在慢性氣道炎癥與結構重塑中的關鍵功能。機制上,KCNJ2通過調控細胞內外離子流動影響NLRP3炎癥小體磷酸化促進其激活,誘導氣道上皮釋放"警報素"來驅動以嗜酸性粒細胞浸潤為特征的2型炎癥反應,導致氣道黏液高分泌與組織重塑。進一步研究發現,靶向抑制KCNJ2能有效阻斷上述信號軸,顯著緩解2型炎癥反應、黏液高分泌、氣道重塑等肺部病理,顯示出良好的疾病治療潛力。該研究深化了對慢性氣道疾病發病機制的理解,鑒定出2型炎癥內型的新靶點與干預候選分子,為開發以離子通道KCNJ2及炎癥小體NLRP3為靶點的治療策略奠定了基礎。
09、靶點:MINDY2
應用:TNF-α介導的炎癥性疾病的潛在治療靶點
來源:Deubiquitinating enzyme MINDY2 regulates TNF-α-induced cell death by targeting RIPK1.Cell Rep,2026 Mar 20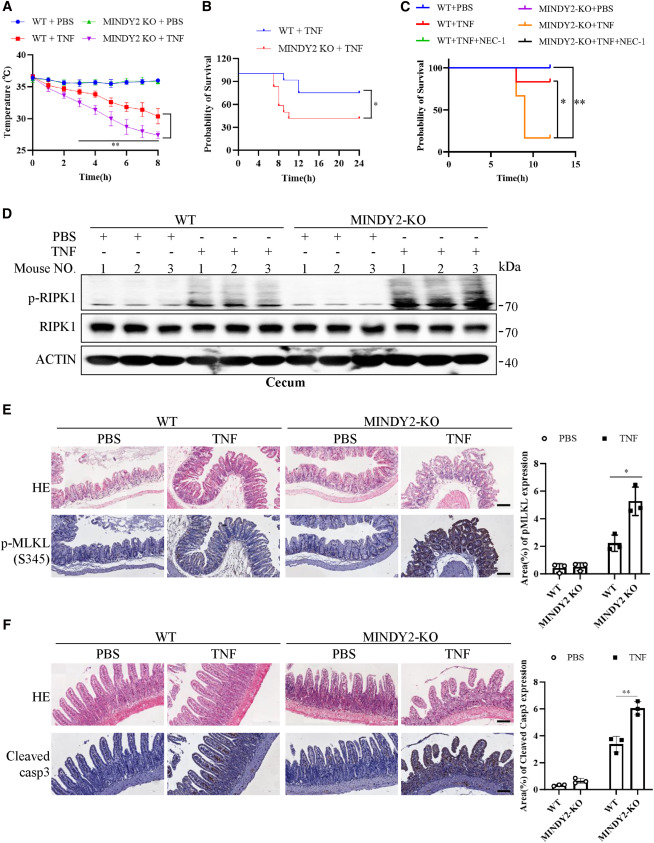
福建醫科大學李立勝團隊和廈門大學林娟團隊在Cell Reports發表研究,系統闡明了去泛素化酶MINDY2調控TNF-α誘導細胞死亡的分子機制。研究發現,MINDY2通過去除RIPK1的泛素化修飾,削弱TNFR1對RIPK1的募集,進而抑制復合物I信號通路及下游RIPK1依賴性凋亡與壞死性凋亡。機制研究表明,MINDY2結合RIPK1并通過其酶活性催化后者去泛素化,通過去除RIPK1 K612位點泛素化來抑制死亡結構域介導的TNFR1-RIPK1相互作用以及RIPK1的同源相互作用。動物實驗表明,MINDY2缺失顯著加重TNF-α誘導的全身炎癥反應綜合征,RIPK1激酶抑制劑Nec-1可有效逆轉上述病理表型,提示MINDY2在體內通過抑制RIPK1依賴性細胞死亡發揮對過度炎癥反應的保護作用。該研究不僅揭示了MINDY2作為RIPK1依賴性細胞死亡關鍵內源性檢查點的新角色,完善了TNF-α信號通路的精細調控網絡,也為TNF-α介導的炎癥性疾病提供了潛在治療靶點。
10、靶點:PHGDH
應用:癌癥的潛在治療靶點
來源:Targeting the noncanonical function of metabolic enzyme PHGDH in driving PD-L1 expression and cancer immune evasion.Cell Rep Med,2026 Apr 21
羅格斯大學癌癥研究所馮朝暉教授團隊與胡文蔚教授團隊在Cell Reports Medicine發表研究,揭示了代謝酶PHGDH在腫瘤中的一項關鍵非經典功能:通過上調免疫檢查點蛋白PD-L1的表達,促進腫瘤免疫逃逸,而且這一過程不依賴其酶活性。代謝重編程在腫瘤發生發展過程中發揮著至關重要的作用,磷酸甘油酸脫氫酶(PHGDH)作為絲氨酸合成通路的首個限速酶,在多種癌癥中高表達,并與不良預后密切相關。近年來研究發現許多代謝酶不僅具有傳統的代謝功能,還具備獨立于酶活性的"非經典功能",但PHGDH在腫瘤免疫中的具體作用及其分子機制仍有待深入研究。研究人員通過基于LC-MS/MS的定量蛋白質組學分析,發現PD-L1是PHGDH敲除后顯著下調。進一步通過Western blot和qPCR證實,在多種癌細胞中PHGDH敲除或敲低后均可顯著降低PD-L1的mRNA和蛋白水平,且該調控不依賴PHGDH的酶活性。機制研究發現,PHGDH可直接結合到RAF1上并促進其活化,從而激活下游MEK/ERK通路上調PD-L1表達。體內實驗顯示,抗PD-1治療在對照組中顯著抑制腫瘤生長,而在PHGDH敲除組中的抑制效果明顯減弱,提示PHGDH高表達的腫瘤更依賴PD-L1介導的免疫逃逸機制,從而對免疫治療更為敏感。鑒于現有的PHGDH抑制劑主要抑制其酶活性而非降解PHGDH蛋白本身,將PHGDH抑制劑與抗PD-1治療聯合使用,可在PHGDH高表達腫瘤中產生更優的抗腫瘤效果。該研究不僅確立了PHGDH作為PD-L1重要調控因子的地位,也為理解其致癌機制提供了新的視角,提示其作為治療靶點的潛在價值,為靶向PHGDH的新型治療策略提供了理論依據。
推薦產品
參考文獻
[1] MTSS1-dependent ubiquitin modifications mediated by FBXO44 remodel the actin cytoskeleton to promote gastric cancer progression.Mol Cancer,2026 Apr 17
應用:胃癌等疾病的潛在治療靶點
來源:MTSS1-dependent ubiquitin modifications mediated by FBXO44 remodel the actin cytoskeleton to promote gastric cancer progression.Mol Cancer,2026 Apr 17
圖源:10.1186/s12943-026-02668-9[1]
中國科學院杭州醫學研究所與浙江省腫瘤醫院覃江江研究員團隊在Molecular Cancer期刊發表研究論文,揭示FBXO44/MTSS1信號軸介導肌動蛋白細胞骨架重塑促進胃癌進展的新機制。研究發現FBXO44通過與MTSS1相互作用調控Rac1核-胞質轉運,通過K63和K11兩種不同的泛素化程序分別促進Rac1入核和誘導MTSS1降解,這種協調機制重塑Rac1信號分布并介導肌動蛋白細胞骨架重構。FBXO44過表達與晚期胃癌Rac1信號增強相關,FBXO44/MTSS1軸表達平衡顯著影響患者預后。該研究為開發新型抗胃癌藥物提供理論基礎,也為靶向微絲細胞骨架的抗腫瘤治療策略提供新靶點。
02、靶點:PI16
應用:HIV治療的潛在靶點
來源:Systematic discovery of pro- and anti-HIV host factors in primary human CD4+ T cells.Cell,2026 Apr 20
圖源:10.1016/j.cell.2026.03.046[2]
Cell近期發表了一篇研究,研究人員在原代人類CD4+ T細胞中通過CRISPRn和CRISPRa全基因組篩選,系統性發現了決定HIV感染命運的宿主因子。研究揭示PI16通過結合Arp2/3復合物和Gaq信號蛋白抑制病毒包膜與細胞膜融合;親環素家族蛋白PPID(Cyp40)與已知的HIV"幫兇"CypA具有62%序列同一性,但PPID通過特有的TPR結構域招募TOM70等宿主分子機器,嚴重阻礙HIV核心的核輸入。研究發現包括H60P、E248K和H146Y在內的多個氨基酸替換可顯著提升人類PPID的抗病毒活性,構建的CypA-CTPR嵌合體也能將促病毒因子CypA改造為強效抗病毒武器。這些發現為開發基于宿主的HIV干預策略提供了重要靶點。
03、靶點:BRD4
應用:
治療肥胖和胰島素抵抗的潛在靶點
來源:BRD4 directs myofiber identity and metabolic adaptation through CHD4 cooperation.Nat Commun,2026 Apr 09
圖源:10.1038/s41467-026-71529-2[3]
南京大學甘振繼教授團隊在Nature Communications發表研究,揭示表觀遺傳"閱讀器"BRD4作為連接營養信號與骨骼肌代謝表型的關鍵樞紐。研究發現BRD4表達水平與受試者BMI呈正相關、與慢肌纖維比例呈負相關,高脂飲食會導致骨骼肌中BRD4蛋白水平顯著上升。骨骼肌特異性敲除BRD4會誘發"快肌向慢肌"重塑,大幅提升線粒體氧化能力和產熱激活,增加全身能量消耗以抵抗肥胖和胰島素抵抗。機制上,BRD4定位結合快肌基因增強子區域,與MEF2及CHD4形成轉錄調控復合物共同維持快肌基因表達。使用BRD4小分子抑制劑JQ1治療不僅顯著降低脂肪含量、改善糖穩態,更重要的是有效保留肌肉質量,為克服GLP-1類藥物伴隨肌肉萎縮的副作用提供了"減脂保肌"的新策略。
04、靶點:SHANK3
應用:自閉癥及其他神經發育障礙的治療靶點
來源:Dynamic recruitment of CaMKII into SHANK3 phase-separated condensates tunes postsynaptic density remodeling during long-term potentiation.Nat Commun,2026 Apr 20
圖源:10.1038/s41467-026-72234-w[41]
中國科學院深圳先進技術研究院張淑雯團隊聯合黃良宇團隊在Nature Communications發表研究,通過液-液相分離概念揭示自閉癥相關SHANK3蛋白調控突觸可塑性的新機制。研究發現SHANK3通過內在無序區域IDR3(尤其是IDR3-3亞區)介導液相分離,全長SHANK3能形成動態凝聚體并強定位至樹突棘,維持靜息態中正常PSD體積和AMPAR表達。突觸激活后,LTP關鍵激酶CaMKII被迅速招募到SHANK3相分離凝聚體中,該過程需要CaMKII自身磷酸化及SHANK3的IDR3-1亞區內RRK基序,進一步加速凝聚體融合與PSD重塑。與人類自閉癥相關的SHANK3突變(InsG3680)導致部分IDR3及SAM結構域缺失,抑制液相分離功能,造成PSD重塑和AMPAR招募缺陷。該研究揭示了活動依賴性神經可塑性的全新分子機制,為自閉癥及其他神經發育障礙的治療靶點提供了新方向。
05、靶點:TLR8
應用:類風濕性關節炎的潛在治療靶點
來源:Toll-like receptor 8 is a therapeutic target in rheumatoid arthritis.Arthritis Rheumatol,2026 Apr 13
圖源:10.1002/art.70175[5]
清華大學尹航課題組聯合協和醫院、北京拓領博泰生物科技有限公司在風濕病學國際期刊Arthritis & Rheumatology發表研究,發現Toll樣受體8(TLR8)是類風濕性關節炎(RA)的關鍵藥物靶點。類風濕性關節炎是高發的慢性自身免疫疾病,2025年全球患病人數約4220萬人,其中中國患者約620萬,目前尚無根治手段。研究團隊發現RA患者體內TLR8高表達且與病情呈正相關性,TLR8通過3條主要途徑推動RA進展:刺激滑膜細胞異常增殖、促進內皮細胞形成新生血管、促使滑膜細胞和免疫細胞釋放更多炎性因子。在臨床前動物模型中,使用TLR8特異性抑制劑進行干預,關節炎臨床評分和腫脹程度顯著下降,關節病理損傷明顯改善。基于該機理開發的TLR8特異性小分子抑制劑目前已進入II期臨床階段并取得優異療效數據,為RA精準治療提供了全新策略。
06、靶點:AFABP/FABP4
應用:治療肥胖相關炎癥性腸病(IBD)的潛在靶點
來源:Obesity impairs gut repair via AFABP-mediated iron overload in intestinal stem cells.Nat Metab,2026 Jan
圖源:10.1038/s42255-025-01425-4[6]
四川大學華西醫院陳海洋教授團隊在Nature Metabolism發表研究,揭示了肥胖損害腸道修復的全新分子機制。研究發現,肥胖時脂肪細胞分泌的脂肪細胞脂肪酸結合蛋白(AFABP/FABP4)會結合血漿轉鐵蛋白(Transferrin, TSF),干擾鐵轉運功能,導致血清非轉鐵蛋白結合鐵(NTBI)升高,引發腸干細胞鐵過載。鐵過載抑制PPARα信號,導致損傷后過氧化物酶體功能下降,進一步擾亂Wnt/Notch分化相關信號平衡,最終阻斷成熟上皮分化,拖慢腸道修復。研究團隊通過Cre依賴AAV在脂肪組織中定點提高AFABP,直接在瘦鼠身上復現了相關表型,為"脂肪-腸道"器官間通訊提供了強有力的因果證據。相反,脂肪特異性敲除AFABP或使用小分子抑制劑BMS309403處理肥胖小鼠,腸道再生能力顯著恢復。基于AFABP-Transferrin-鐵-過氧化物酶體軸,使用鐵螯合劑DFO或過氧化物酶體/PPAR激活劑(如非諾貝特、NaPB或吡格列酮),均可有效逆轉肥胖小鼠的腸道修復缺陷,為治療肥胖相關炎癥性腸病(IBD)提供了極具潛力的可成藥策略。
07、靶點:BDNF
應用:肌萎縮側索硬化癥(ALS,漸凍癥)的潛在治療靶點
來源:BDNF insufficiency exacerbates ALS progression.Cell Rep Med,
2026 May 19
圖源:10.1016/j.xcrm.2026.102758[7]
清華大學魯白教授、復旦大學腦科學轉化研究院袁鵬青年研究員、北京大學第三醫院樊東升教授作為共同通訊作者在Cell子刊Cell Reports Medicine發表研究,證實腦源性神經營養因子(BDNF)不足會加劇肌萎縮側索硬化癥(ALS,漸凍癥)的進展。研究發現,人類BDNF基因中常見的單核苷酸多態性rs6265導致Val66Met突變,降低BDNF分泌量,從而縮短ALS患者的生存期。利用ALS致病基因FUS R521C敲入小鼠模型進一步證明,BDNF半劑量不足會導致壽命縮短、運動功能障礙加速以及運動神經元死亡加劇。重要的是,使用激動性抗體激活BDNF受體TrkB,能夠有效挽救ALS相關表型,在其他ALS小鼠模型中,TrkB激活抗體也顯示出優于目前ALS治療藥物利魯唑的治療效果。該研究表明BDNF不足可能是ALS進展的關鍵促成因素,激活BDNF-TrkB通路代表了一種有前景的ALS治療新策略。
08、靶點:KCNJ2
應用:慢性氣道疾病的潛在治療靶點
來源:KCNJ2 is required for NLRP3 inflammasome activation that drives allergic airway inflammation and remodeling.Adv Sci (Weinh),2026 May
圖源:10.1002/advs.202517666[8]
廣州國家實驗室與廣州醫科大學殷文廣、冉丕鑫、李時悅團隊在Advanced Science期刊發表研究,鑒定出慢性氣道疾病新干預靶點KCNJ2,闡明了其驅動2型炎癥反應與氣道重塑的分子機制。2型炎癥為慢性氣道疾病一主要內型,約20%-40%慢阻肺患者、50%-70%哮喘患者與2型炎癥有關。內向整流型鉀離子通道作為鉀離子通道家族中的主要類型,在穩定細胞膜電位中發揮主要作用。研究從臨床患者數據與樣本分析出發,結合動物模型、細胞、類器官培養等,發現了KCNJ2在慢性氣道疾病發生中的作用,提出了"鉀離子通道-炎癥小體-警報素-2型炎癥反應"信號軸調控模型,揭示了其在慢性氣道炎癥與結構重塑中的關鍵功能。機制上,KCNJ2通過調控細胞內外離子流動影響NLRP3炎癥小體磷酸化促進其激活,誘導氣道上皮釋放"警報素"來驅動以嗜酸性粒細胞浸潤為特征的2型炎癥反應,導致氣道黏液高分泌與組織重塑。進一步研究發現,靶向抑制KCNJ2能有效阻斷上述信號軸,顯著緩解2型炎癥反應、黏液高分泌、氣道重塑等肺部病理,顯示出良好的疾病治療潛力。該研究深化了對慢性氣道疾病發病機制的理解,鑒定出2型炎癥內型的新靶點與干預候選分子,為開發以離子通道KCNJ2及炎癥小體NLRP3為靶點的治療策略奠定了基礎。
09、靶點:MINDY2
應用:TNF-α介導的炎癥性疾病的潛在治療靶點
來源:Deubiquitinating enzyme MINDY2 regulates TNF-α-induced cell death by targeting RIPK1.Cell Rep,2026 Mar 20
圖源:10.1016/j.celrep.2026.117134[9]
福建醫科大學李立勝團隊和廈門大學林娟團隊在Cell Reports發表研究,系統闡明了去泛素化酶MINDY2調控TNF-α誘導細胞死亡的分子機制。研究發現,MINDY2通過去除RIPK1的泛素化修飾,削弱TNFR1對RIPK1的募集,進而抑制復合物I信號通路及下游RIPK1依賴性凋亡與壞死性凋亡。機制研究表明,MINDY2結合RIPK1并通過其酶活性催化后者去泛素化,通過去除RIPK1 K612位點泛素化來抑制死亡結構域介導的TNFR1-RIPK1相互作用以及RIPK1的同源相互作用。動物實驗表明,MINDY2缺失顯著加重TNF-α誘導的全身炎癥反應綜合征,RIPK1激酶抑制劑Nec-1可有效逆轉上述病理表型,提示MINDY2在體內通過抑制RIPK1依賴性細胞死亡發揮對過度炎癥反應的保護作用。該研究不僅揭示了MINDY2作為RIPK1依賴性細胞死亡關鍵內源性檢查點的新角色,完善了TNF-α信號通路的精細調控網絡,也為TNF-α介導的炎癥性疾病提供了潛在治療靶點。
10、靶點:PHGDH
應用:癌癥的潛在治療靶點
來源:Targeting the noncanonical function of metabolic enzyme PHGDH in driving PD-L1 expression and cancer immune evasion.Cell Rep Med,2026 Apr 21
圖源:10.1016/j.xcrm.2026.102704[10]
羅格斯大學癌癥研究所馮朝暉教授團隊與胡文蔚教授團隊在Cell Reports Medicine發表研究,揭示了代謝酶PHGDH在腫瘤中的一項關鍵非經典功能:通過上調免疫檢查點蛋白PD-L1的表達,促進腫瘤免疫逃逸,而且這一過程不依賴其酶活性。代謝重編程在腫瘤發生發展過程中發揮著至關重要的作用,磷酸甘油酸脫氫酶(PHGDH)作為絲氨酸合成通路的首個限速酶,在多種癌癥中高表達,并與不良預后密切相關。近年來研究發現許多代謝酶不僅具有傳統的代謝功能,還具備獨立于酶活性的"非經典功能",但PHGDH在腫瘤免疫中的具體作用及其分子機制仍有待深入研究。研究人員通過基于LC-MS/MS的定量蛋白質組學分析,發現PD-L1是PHGDH敲除后顯著下調。進一步通過Western blot和qPCR證實,在多種癌細胞中PHGDH敲除或敲低后均可顯著降低PD-L1的mRNA和蛋白水平,且該調控不依賴PHGDH的酶活性。機制研究發現,PHGDH可直接結合到RAF1上并促進其活化,從而激活下游MEK/ERK通路上調PD-L1表達。體內實驗顯示,抗PD-1治療在對照組中顯著抑制腫瘤生長,而在PHGDH敲除組中的抑制效果明顯減弱,提示PHGDH高表達的腫瘤更依賴PD-L1介導的免疫逃逸機制,從而對免疫治療更為敏感。鑒于現有的PHGDH抑制劑主要抑制其酶活性而非降解PHGDH蛋白本身,將PHGDH抑制劑與抗PD-1治療聯合使用,可在PHGDH高表達腫瘤中產生更優的抗腫瘤效果。該研究不僅確立了PHGDH作為PD-L1重要調控因子的地位,也為理解其致癌機制提供了新的視角,提示其作為治療靶點的潛在價值,為靶向PHGDH的新型治療策略提供了理論依據。
推薦產品
| 靶點 | 重組蛋白 | 貨號 |
| BDNF | Recombinant Human Brain-derived neurotrophic factor (BDNF) | CSB-EP002655HU |
| BRD4 | Recombinant Human Bromodomain-containing protein 4 (BRD4), partial | CSB-EP002802HU |
| FABP4 | Recombinant Human Fatty acid-binding protein, adipocyte (FABP4) | CSB-EP007945HU |
| FBXO44 | Recombinant Human F-box only protein 44 (FBXO44) | CSB-EP872497HU |
| KCNJ2 | Recombinant Human Inward rectifier potassium channel 2 (KCNJ2), partial | CSB-MP012055HU1 |
| MINDY2 | Recombinant Mouse Ubiquitin carboxyl-terminal hydrolase MINDY-2 (Mindy2) | CSB-EP750855MO |
| PHGDH | Recombinant Human D-3-phosphoglycerate dehydrogenase (PHGDH), partial | CSB-EP017920HU1 |
| PI16 | Recombinant Human Peptidase inhibitor 16 (PI16), partial | CSB-MP017951HU |
| SHANK3 | Recombinant Human SH3 and multiple ankyrin repeat domains protein 3 (SHANK3), partial | CSB-EP880134HU |
| TLR8 | Recombinant Human Toll-like receptor 8 (TLR8), partial | CSB-YP023607HU |
參考文獻
[1] MTSS1-dependent ubiquitin modifications mediated by FBXO44 remodel the actin cytoskeleton to promote gastric cancer progression.Mol Cancer,2026 Apr 17
[2]Systematic discovery of pro- and anti-HIV host factors in primary human CD4+ T cells.Cell,2026 Apr 20
[3]BRD4 directs myofiber identity and metabolic adaptation through CHD4 cooperation.Nat Commun,2026 Apr 09
[4]Dynamic recruitment of CaMKII into SHANK3 phase-separated condensates tunes postsynaptic density remodeling during long-term potentiation.Nat Commun,2026 Apr 20
[5]Toll-like receptor 8 is a therapeutic target in rheumatoid arthritis.Arthritis Rheumatol,2026 Apr 13
[6]Obesity impairs gut repair via AFABP-mediated iron overload in intestinal stem cells.Nat Metab,2026 Jan
[7]BDNF insufficiency exacerbates ALS progression.Cell Rep Med,2026 May 19
[8]KCNJ2 is required for NLRP3 inflammasome activation that drives allergic airway inflammation and remodeling.Adv Sci (Weinh),2026 May
[9]Deubiquitinating enzyme MINDY2 regulates TNF-α-induced cell death by targeting RIPK1.Cell Rep,2026 Mar 20
[10]Targeting the noncanonical function of metabolic enzyme PHGDH in driving PD-L1 expression and cancer immune evasion.Cell Rep Med,2026 Apr 21
*免責聲明:華美生物內容團隊僅是分享和解讀公開研究論文及其發現,本文僅作信息交流,文中觀點不代表華美生物立場,請理解。
下一篇: 最后一頁















